|
Developing Saccharomyces cerevisiae RAS G19C for Modeling Human KRAS G12C Cancers Samuel Barnett, University of Maryland, Baltimore County; Howard Community College Mentored by: Joseph C. Sparenberg |
Abstract
Cancer is characterized by uncontrolled cell growth due to mutations and is often caught in later stages. One such mutation in humans is in the oncogene KRAS. The protein translated from KRAS regulates cell growth, and mutations are implicated in cancer. GTP is the substrate for the KRAS protein, which activates the protein leading to cell replication. When GTP is converted to GDP, KRAS is inactivated. Mutations in KRAS can prevent GTP to GDP conversion leading to unregulated proliferation. We are using Saccharomyces cerevisiae (baker’s yeast) cells, due to their similarities to human cells (30-40% genetically identical) and transferable methods. We propose a method to model a commonly studied mutation, the glycine to cysteine substitution at the 12th amino acid in KRAS-based cancer (KRASG12C) in humans using S. cerevisiae by modifying RAS1 (an analog to KRAS). Modifications of the RAS1 gene leading to a mutation in the subsequent protein may disrupt signaling by restricting the RAS1 protein from deactivating, leading to rapid cell proliferation (analogous to KRAS tumorigenesis). Electrophoresis results indicated a custom plasmid is required. The RAS1 and MS2 encoding regions within the custom plasmid can lead to direct transfection and visualization with a complementary tagging plasmid. Our model could visualize a live mRNA’s life cycle and potentially predict cancerous tumor growth. Using S. cerevisiae cells as a carcinogenesis model may elucidate inhibitors to potentially create an on/off switch for replication related to the KRAS G12C mutation.
Introduction
Carcinogenesis is the formation of a tumor(s) through unregulated cell proliferation caused by environmental factors and genetic irregularities. These factors negatively impact an organism in several aspects. Potential interference of the cell cycle because of genetic alterations in proto-oncogenes and tumor repressor genes can result in carcinogenesis. These genes are vital in the cell cycle because the encoded proteins determine cell replication. Proto-oncogenes promote cell growth, while tumor repressor genes limit cell growth based on cell signaling. Mutations or alterations within proto-oncogenes result in uncontrolled cell growth because they are “reprogrammed” to promote cell growth constitutively. Mutations to tumor suppressor genes result in cancer because the subsequent protein’s ability to stop cell proliferation is downregulated or “knocked out.” Numerous proto-oncogenes and tumor suppressor genes are located in different cell cycle stages and involved in biochemical pathways that create small molecule intermediates based on the cell’s needs or fate.
The cell cycle comprises four stages; G1, S (interphase), G2, and mitosis. Different processes occur within the G1, S, and G2 phases to prepare for cell division within the mitotic phase. The mitogen-activated protein kinase pathway (MAPK) MAPK pathway is a significant biochemical pathway involving cell signaling cascades responsible for entities that enable the processes within the cell cycle to occur [7]. One important part of the MAPK pathway involves the KRAS gene (a proto-oncogene) [7].
KRAS’s specific role has been extensively researched. The protein sequenced by the KRAS gene is activated by RAS guanine exchange factors (RASGEFS) that ultimately allow cell growth through downstream effectors (cell growth initiation factors)[7]. The MAPK pathway helps control cell growth by cycling native KRAS proteins between the guanosine triphosphate (GTP) bound “on” and guanosine diphosphate (GDP) “off” states based on the cell’s needs. Certain mutations cause a cell to constantly be in the “on” state [1,6]. Mutations to glycine 12 (G12), glycine 13 (G13), or glutamine 16 (Q16) to the KRAS gene prevent the subsequent protein from deactivating due to the tightly bound GTP substrate [7]. Overactive KRAS proteins cause unregulated cell proliferation, tumor growth, and subsequent metastasis [1,6].
- cerevisiae is a promising model organism for representing KRAS-based cancer within a living system because of its low cost and ease of use. Analogous structures of the S. cerevisiae RAS1 and the human KRAS proteins confirmed S. cerevisiae is an effective organism for visualizing KRAS cancers [1]. Downstream effectors are also highly conserved among the KRAS and RAS1 genes [6]. Specifically, the MAPK pathway, commonly implicated in KRAS-based cancer growth, is orthologous in S. cerevisiae. Resistance to treatments after a prolonged period (acquired resistance) is a significant challenge [5].
The KRAS protoncogene is susceptible to acquired resistance and mutations and is responsible for one million deaths annually [1,5]. Trials for treatments for KRAS-based cancers have been proven temporarily effective but are promising [4]. In clinical trials, the FDA-approved KRAS-targeting drug Sotorasib allows moderate (12 months) survival [4]. The efficacy of drugs like Sotorasib could be improved by performing preliminary research within a more cost-effective representative living system using a suitable model organism like S. cerevisiae.
Tucci, et al. analyzed a S. cerevisiae gene expression using an mRNA tagging system utilizing an encoding and tagging plasmid [5]. We introduce a similar method to model a commonly studied cancerous mutation in the KRAS protein (Fig. 1), the glycine to cysteine substitution at the 12th amino acid (KRASG12C) in human cells using S. cerevisiae [2,3]. The RAS1 S. cerevisiae protein overlaps with KRASG12 at the 19th position meaning KRASG12C would be analogous to RAS1G19C [1]. KRASG12C is historically difficult to treat because of the subsequent proteins’ smooth structure (limited binding pockets) but has been counteracted by Sotorasib [1,3,4,5,6]. Using RAS1G19C as a screening model for mutative cancers may elucidate KRAS therapeutics in a living system to measure their efficacy.
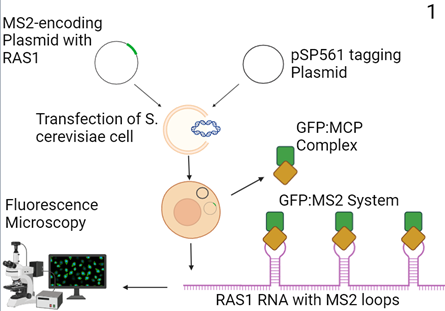
Figure 1: Proposed Molecular cloning model (3). A custom MS2-encoding plasmid with an adjacent RAS1-encoding region and a complementary tagging plasmid (pSP561) is transfected into S. cerevisiae cells. The transcribed custom plasmid encodes RAS1 RNA with MS2 stem-loops and the pSP561 plasmid creates a green fluorescent protein (GFP):MS2-coating protein (MCP) complex that binds to the loops. Lucent signals are then assessed using fluorescence microscopy.
We would need to analyze corresponding proteins or genes to determine if S. cerevisiae is a viable model for specific conditions that contain a mutation. If S. cerevisiae is an incompatible organism, other Saccharomyces species could be compatible with our methods because of the diversity of similar genes and structures in different species [7].
Methods
Media: Super optimal broth (SOB) and yeast peptone dextrose broth (YPD) media were ordered from Thermofisher Scientific.
Preparation of E. coli plates: Media comprises 2.5g Tryptone, 1.25g yeast extract, 2.5g sodium chloride, 3.75g of agar, 250 ml of autoclaved deionized water, and specified bacterial resistant agent. Media was autoclaved and then poured into plates until visually full and dried for 20 minutes to 24 hours. About 50µL of E. coli XL-1 Blue cells ordered from Agilent were spread onto plates using sterile cell spreaders and incubated at 37°C overnight.
Preparation of S. cerevisiae plates: Liquid media used to create plates are composed of 5.0 grams of sodium chloride, 10.0 grams of agar, 2.5 grams yeast extract, 5.0 grams tryptone, and autoclaved deionized water was added to reach a total volume of 1000 mL. Media was assessedinitial pH and NaOH was added until pH 7 was reached. The mixture was autoclaved, cooled, and poured into Petri dishes to solidify. Roughly 200µL of S. cerevisiae cells were spread onto the plates using sterile cell spreaders and incubated at 37 °C overnight.
Bacterial Transformation: 50ng of Circular DNA vectors (plasmids) are inserted into 100µL of Escherichia coli XL-1 Blue cells. Insertion occurs through a 42°C water bath heat shock for 45 seconds and usage of 1.7µL ꞵ-mercaptoethanol (BME). The transformed E. coli is allowed to grow in a shaker incubator overnight. After incubation, the 5µL of transformed E. coli is plated and incubated at 37°C overnight. Transformed E. coli is extracted from the plate using a pipette tip, grown in SOB media, and purified using the GeneJET™ Plasmid Miniprep Kit.
Polymerase Chain Reaction: 1µL forward primer, 1µL reverse primer, 0.25µL Taq polymerase, 5µL 10x Tris-HCl Taq PCR buffer, 1µl of dNTP, 1µl of MgCl2, and purified RAS1 plasmid (pBY011) from E. coli XL-1 Blue that is volume dependent based on measured concentration to achieve 50 ng of DNA were added to separate PCR tubes for amplification and isolation of the RAS1 gene. Water is added to the final volume of 25µL. Our profile details a denaturation of 95°C, an annealing of 64°C, and an elongation of 72°C. Purification of PCR product followed the Thermofisher Scientific GeneJET™ PCR Purification Kit.
Primer Design: We used the SnapGene program to analyze the plasmid maps of the pSP568 plasmid. Direct DNA analysis was performed using a custom code directly comparing DNA sequence data from the pBY011 and pSP568 plasmids.
Gel electrophoresis: A 1.5% agarose gel solution is made with 1.5g of agarose and 100mL of 1x tris-acetate-EDTA. The solution is then microwaved for roughly 60 seconds, poured into a casting tray in the gel electrophoresis apparatus, and allowed to sit for 1 hour. ~1000mL 1x tris-acetate-EDTA (TAE) buffer is poured into the electrophoresis chamber for an electrical current at 120 amp to run the gel. After running the gel was immediately allowed to sit in a 1:10000 SYBR™ Green:TAE gel stain solution for approximately 30 minutes. Gel analysis was performed using the GelDoc imager.
pSP561 Plasmid: pSP561 was a gift from Serge Pelet (Addgene plasmid # 139497 ; http://n2t.net/addgene:139497 ; RRID:Addgene_139497)
pSP568 Plasmid: pSP568 was a gift from Serge Pelet (Addgene plasmid # 139493 ; http://n2t.net/addgene:139493 ; RRID:Addgene_139493)
pBY011 Plasmid: Plasmid was ordered from DNASU (plasmid ID: ScC000095575)
Custom Plasmid Creation: Custom RAS1-MS2-SL plasmid was formulated and ordered by Addgene
Results
The viability of plasmids for future experimentation was assessed using bacterial transformation. Wild-type E. coli lacks innate resistance to ampicillin. Transformation with ampicillin-resistant plasmids essential for the fluorescing of RAS1 RNA (pBY011, pSP568, pSP561) visually displayed growth greater than the wild-type control on ampicillin plates (Figure 2).
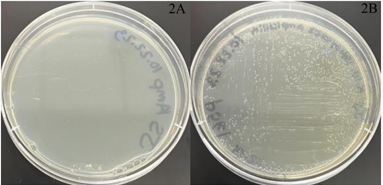
Figure 2A and 2B: Transformation results with negative and positive control plasmid growths. The picture on the left contains wild-type untransformed E. coli cells (negative control – 2A). The pSP561-transformed E. coli exhibits the ampicillin-resistant gene in the plasmid and shows cell growth (positive control – 2B).
Electrophoresis validated no present DNA from the RAS1 plasmid (pBY011) meaning the plasmids had to be reassessed (Figure 3). Upon analysis of the primers used for the PCR process, primers appeared to be binding on themselves. Primers for this process were designed to be specific to our pBY011 plasmid for isolating the RAS1 gene. Other primer candidates were unlikely after plasmid map analysis.

Figure 3: Isolated and purified RAS1 from PCR was pipetted into the gel along with a ladder. The ladder depicted in the middle lane is alone in gel with no other DNA, indicating no DNA or too little DNA in the run.
Direct sequence comparisons revealed the pSP568 plasmid is not viable for RAS1-MS2 conjoining due to the lack of homologous sequences for restriction digest. Plasmid maps and direct sequences revealed no regions for RAS1 to be inserted properly with alternative primer designs. A custom plasmid with an MS2-encoding region adjacent to the RAS1-encoding region was deemed necessary and ordered for future experimentation (Figure 4).
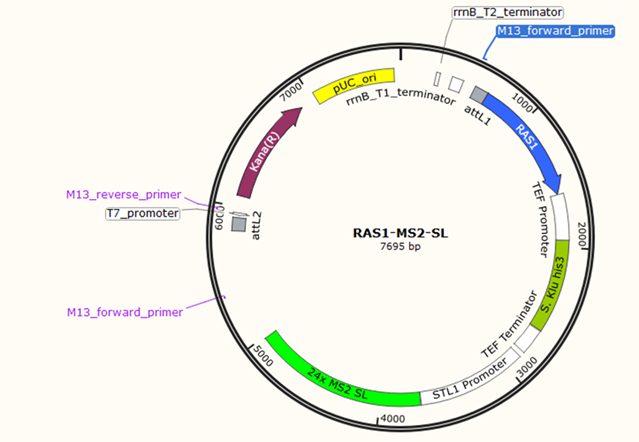
Figure 4: Custom RAS1-MS2-stem-loop (RAS1-MS2-SL) plasmid ordered from Thermofisher Scientific. The RAS1 gene is depicted in the plasmid with the MS2-encoding region directly adjacent to the major STL1 promoter. Kana(R) is a gene that enables kanamycin resistance in cells that uptake the plasmid.
Discussions and Conclusion
Two plasmids were confirmed inviable: the RAS1-encoding pBY011 plasmid and the MS2-encoding pSP568 plasmid. The primers used in the PCR process to isolate the RAS1 gene from the pBY011 plasmid bound to themselves and subsequent primer designs were unlikely due to the inflexibility of primer sites on the plasmid. We considered isolating the MS2-stem-loop (MS2-SL) encoding region for cloning into the pBY011 plasmid but primers would interfere with either the MS2 encoding or promoter region. The MS2 encoding region is required to be directly adjacent to the major promoter region on the plasmid to ensure the complex is transcribed with the loops.
Confirmation of plasmids by bacterial transformation with an ampicillin-resistant gene validated plasmid viability and rendered a stock of the pSP561 green fluorescent protein (GFP): MS2 coating protein (MCP) plasmid for transfection into S. cerevisiae cells. The kanamycin-resistant Kana(R) gene in RAS1-MS2-SL is present as opposed to the ampicillin-resistant Amp(R) gene in pSP561 indicating the need for kanamycin plates for RAS1-MS2-SL transformed cells.
With a complementary tagging plasmid to our custom plasmid, light signals from the GFP:MS2 SL complex will be viewed in vivo using fluorescence microscopy. Once RAS1 mRNA is visualized, site-directed mutagenesis by PCR is used to induce the human analog G19C mutation. The mutation can initiate and predict cancerous growth meaning inhibitors can be screened for potential drug candidates. Future collaborations could produce promising results in human cell lines.
The inception of cell differentiation to tissue is a widely debated subject, but if cancer can be induced in a single-celled organism like S. cerevisiae, this project may aid in the understanding of why cancer exists.
Acknowledgments
We would like to dedicate this paper and subsequent work to our previous lab member Rayhan Stewart, who tragically passed away earlier this year. Additionally, thanks to the Howard Community College research faculty and the Barry Goldwater Scholarship Foundation for recognizing and supporting our project.
Contact: sbarnet1@umbc.edu, idrees.chaudry@howardcc.edu, katrina.william@howardcc.edu, jsparenberg@howardcc.edu
References
[1] A. A. Doud, et. al. “Is Saccharomyces cerevisiae a Viable Model for Studying Cancer Mutations in KRAS?,” Journal of Research in Progress, vol. 6, no. 1, p. 6, 2023,
[2] E. Tutucci, et. al., “An improved MS2 system for accurate reporting of the mRNA life cycle” Nature Methods, vol. 15, p. 81, 2018, doi:10.1038/nmeth.4502
[3] G. Cazzanelli, et. al. “The Yeast Saccharomyces cerevisiae as a Model for Understanding RAS Proteins and their Role in Human Tumorigenesis,” Cells, vol. 7, no. 2, p. 14, 2018, doi: 10.3390/cells7020014
[4] G. K. Dy, et. al., “Long-Term Outcomes and Molecular Correlates of Sotorasib Efficacy in Patients With Pretreated KRAS G12C-Mutated Non–Small-Cell Lung Cancer: 2-Year Analysis of CodeBreaK 100,” Journal of Clinical Oncology, vol. 41, no. 18, p. 3311, 2023, doi: 10.1200/JCO.22.02524
[5] L. M. Mustachio, et. al., “Targeting KRAS in Cancer: Promising Therapeutic Strategies,” Cancers, vol. 13, no. 6, p. 1204, 2021, doi: 10.3390/cancers13061204
[6] L. Vanderwaeren, et. al. “Saccharomyces cerevisiae as a Model System for Eukaryotic Cell Biology, from Cell Cycle Control to DNA Damage Response,” International Journal of Molecular Sciences, vol. 23, no. 19, p. 11665, 2022, doi: 10.3390/ijms231911665
[7] M. Cargnello and P. P. Roux, “Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases,” Microbiology and Molecular Biology Reviews, vol. 75, no. 1, pp. 50–83, Mar. 2011, doi: 10.1128/mmbr.00031-10.
[8] N. Seyidoglu and C. Aydin “Saccharomyces: Is a Necessary Organism or a Biological Warrior” IntechOpen: Saccharomyces, 2021 , doi: 10.5772/intechopen.96029
[9] Wosika V. and Pelet S., “Single-particle imaging of stress-promoters induction reveals the interplay between MAPK signaling, chromatin and transcription factors” Nature Communications, vol. 11, no. 1, pp. 3171, Jun. 23, 2020, doi: 10.1038/s41467-020-16943-w