9 Using CRISPR-Cas9 to Treat Sickle Cell Disease
Karis Weisgerber, Howard Community College
Mentored by: Kathryn S. Jones, Ph.D.
Abstract
Sickle cell disease is caused by a mutation in the gene that encodes a protein in hemoglobin called the beta chain. Hemoglobin is the part of red blood cells that carries oxygen from the lungs to other parts of the body. Sickle cell disease is characterized by red blood cells that are sickle shaped (like a crescent moon), rather than the round shape of healthy red blood cells. These sickle cells get caught on blood vessel walls and can lead to moments of intense pain. Current treatments for this disease are limited, and each contains serious risks. In recent years, gene therapy has become a new potential treatment for sickle cell disease. One specific form of gene therapy, incorporating the use of CRISPR-Cas9 technology, has shown significant promise in clinical trials. This technology functions by editing specific base pairs in DNA and has the ability to fix the mutation causing sickle cell disease. Studies have shown that, in addition to sickle cell disease, this approach could be used to successfully treat many other genetic diseases.
Paper
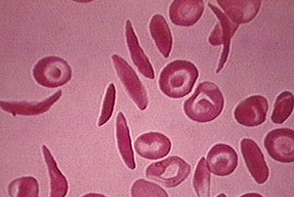
Sickle cell disease is one of the most common disorders caused by a mutation in a single gene, affecting millions of individuals around the world. There are approximately 100,000 sickle cell disease patients in the United States, and one in every 365 African American babies is born with this disease [1]. The disease is characterized by a malfunction in a protein called hemoglobin, the component of red blood cells that is responsible for carrying oxygen throughout the body. Individuals with this disease produce a defective version of hemoglobin called hemoglobin S, rather than the normal version (hemoglobin A) [2]. The mutated hemoglobin precipitates in red blood cells as opposed to being soluble, and this leads to red blood cells that are crescent (sickle) shaped instead of the normal round shape [3]. The image in Figure 1 shows a sample of red blood cells from a carrier of sickle cell disease, with both types of red blood cells. The round cells are the red blood cells with hemoglobin A, and the thin sickle cells have the mutant hemoglobin S.
This genetic disorder is caused by a point mutation (a change of one nucleotide) in the DNA of the gene coding for one of the proteins in hemoglobin (the beta chain). Specifically, a thymine (T) is present in the position where there should be an adenine (A) [5] in the sixth codon of the hemoglobin-beta (HBB) gene on chromosome 11. That mutation causes the amino acid valine to be produced instead of glutamic acid, and this causes irregularity in both the shape and function of the protein. The version of this gene containing the mutation is often referred to as the sickle hemoglobin (HbS) gene because of the hemoglobin S that it encodes. The disease is inherited autosomal recessively: the mutation must be present in both copies of the gene for the individual to have sickle cell disease [3].
The sickle shape of the red blood cells can cause them to get stuck on blood vessel walls, build up, and eventually break down prematurely. This can lead to bacterial infections and strokes. The lack of healthy red blood cells (anemia) can lead to an insufficient amount of oxygen being delivered to vital organs [2]. Vaso-occlusive crises (when circulation is blocked to a certain organ) often occur in sickle cell disease patients because of the sickle cell buildup in the vessels. This leads to a lack of oxygen in that organ and causes a period of excruciating pain [6].
Treatments for this disease are limited. Blood transfusions are performed to alleviate some of the symptoms temporarily, and therapies to prevent the side effects of sickle cell disease such as penicillin prophylaxis, pneumococcal vaccination, and hydroxyurea therapy have been performed. However, these do not cure the disease, and some patients do not react well to these treatments. The only current long-term treatment available is an allogeneic hematopoietic stem cell transplant. Unfortunately, over 80% of patients are not able to find a compatible donor, and individuals who do receive such transplants are at risk for complications such as graft rejection and graft versus host disease [7], both of which can be fatal.
One potentially new treatment for sickle cell disease that seems promising is gene therapy. Gene therapy is the process of introducing genetic material into cells with the intention of causing some sort of therapeutic effect. These effects may include fixing a mutated gene, inserting missing genetic information, or providing the genetic instructions for that cell to perform a different function. One of the most common methods of gene therapy is inserting a viral vector: a virus whose DNA has been edited to incorporate a specific gene, into the cell to transfer that gene into the cell’s genome [8]. Another method of gene therapy involves inserting synthetic oligonucleotides into the target cells to inactivate certain genes. Synthetic oligonucleotides are short strands of DNA or RNA that are engineered to be complementary to the DNA of the particular gene to be turned off. When inserted into the cell, they bind to the DNA of that gene and prevent it from being transcribed into mRNA, ultimately preventing it from producing a protein, and thus silencing its effects. A third method of gene therapy involves inserting mRNA into the cell to be translated by ribosomes and produce a specific protein [9].
Each of these methods comes with serious risks. For example, inserting viral DNA containing a certain gene into the cell’s genome can lead to negative side effects, including cancer. Another negative side effect is immunogenic toxicity. Since the viral vector is foreign to the body, it sometimes can generate an immune response including antibodies to fight the virus. This could ultimately prevent the wanted gene from being inserted into the cell’s genome, and could also lead to adverse effects, such as an allergic reaction. Both side effects could end up causing more problems than the gene therapy was initially aiming to fix [10]. Recently, a radical new form of gene therapy without such side effects has been developed. This form is cheaper and easier to use than others, and it incorporates CRISPR-Cas9.
CRISPR stands for “clustered regularly interspaced short palindromic repeats,” and Cas9 stands for “CRISPR associated protein 9.” The gene therapy developed is a modification of the naturally occurring system found in bacteria called CRISPR-Cas. It was first discovered in a type of bacteria called Escherichia coli in 1987, but its purpose was not discovered until two decades later [8]. This system of alternating repeated DNA sequences and non-repeating DNA sequences is a system of defense that bacteria use against infection from a certain type of virus that infects bacteria called a bacteriophage. When a bacteriophage infects a bacterial cell, the cell inserts DNA homologous to a portion of the bacteriophage’s genome into the CRISPR sequences. If the cell is infected again in the future by the same bacteriophage, it will recognize its foreign DNA from the sequence previously added to the CRISPR array and will be able to fight off the infection [11]. Figure 2 gives a visual depiction of a CRISPR array.
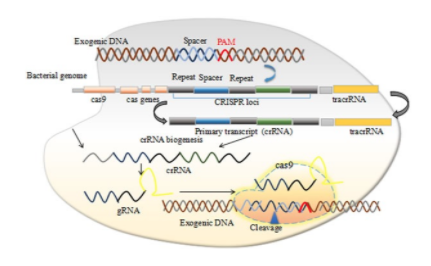
There are multiple different systems of CRISPR-Cas, incorporating different Cas proteins and types of RNA. One specific system of CRISPR-Cas called the Type II system incorporates Cas9 and a type of RNA called trans-activating crRNA (tracrRNA). TracrRNA is complementary to the palindromic repeating sequences of DNA, as named in the acronym “CRISPR.” It is also partially complementary to another kind of RNA called CRISPR RNA, or crRNA. This allows it to bind to crRNA, and the two types together form a duplex RNA. The Cas9 protein is an endonuclease, an enzyme that “cuts” DNA by cleaving the phosphodiester bonds that join nucleotides together. This protein cleaves the DNA of bacteriophages that infect the bacterial cell so that the cell is not harmed by the viruses. The Cas9 protein uses the tracrRNA/crRNA duplex as its guide to tell it which DNA to cut and where to make the cut [11]. Figure 2 shows the mechanism of this process.
In 2012, scientists discovered that this CRISPR-Cas system could be used to cleave a specific sequence of DNA by fusing the tracrRNA/crRNA complex into one single RNA. They could join the crRNA to the tracrRNA, and then engineer the new RNA molecule to be complementary to essentially any target DNA sequence for modification. This engineered RNA molecule would later be known as guide RNA (also referred to as sgRNA or gRNA). This approach initially demonstrated that CRISPR-Cas9 could be used to induce targeted mutations in bacterial genomes, and to activate or inactivate the transcription of specific bacterial genes. Scientists continued to modify CRISPR-Cas9 and it was later demonstrated that it could also be used to alter genes in yeast, certain plants, mice, and cultured human cells [11].
The studies showing that CRISPR-Cas9 could be used to edit the genome of eukaryotic cells, including human cells, raised the possibility that it could be used as a new form of gene therapy. Scientists reasoned that it could be used to correct genetic defects in human genes by site-specific gene editing, and that it would be simpler and more efficient than the approaches in use at the time [10]. In addition, since the engineered gRNA is inserted into the cells without viral proteins, this approach would be less likely to initiate an immune response than traditional gene therapy involving viral vectors.
In 2016, scientists successfully used CRISPR to deactivate the gene that produces a protein involved in cancer development (an immune checkpoint inhibitor called PD-1) in a patient with lung cancer [8]. In this study, they introduced CRISPR-Cas9 with a gRNA targeting the PD-1 gene into lymphocytes (a type of immune cells) isolated from the individual with lung cancer. They then put the genetically modified lymphocytes back into the individual, with the goal of causing the body to attack tumor cells more efficiently, and the PD-1 gene was successfully silenced in the individual’s cells [8].
Later, scientists investigated whether this method could, in addition to simply deactivating genes, be used to correct inherited genetic mutations that cause disease [8]. In the same year, CRISPR-Cas9 was successfully used in cells cultured in a laboratory to correct a point mutation to the SERPINA1 gene that causes a condition called alpha-1 antitrypsin deficiency, characterized by symptoms affecting the lungs and liver [12]. Since then, CRISPR technology has been successfully applied in treating cells with certain cancers, such as lung cancer, breast cancer, and thyroid cancer. It has also been used to treat cells with genetic diseases such as beta-thalassemia, Huntington disease, muscular dystrophy, and Alzheimer’s, and the results have been promising [13]. These initial studies suggest that this technology could be used clinically to treat a wide variety of genetic disorders and other types of conditions caused by changes in the sequences or regulation of genes. These findings were so groundbreaking that the Nobel Prize in Chemistry 2020 was awarded to scientists Emmanuelle Charpentier and Jennifer Doudna for discovering how CRISPR can be programmed to edit DNA in a specific location with guide RNA [14].
Sickle cell disease is one of the diseases that scientists wanted to attempt to treat using CRISPR-Cas9. Initially, scientists wanted to try to change the mutation in the beta-globin gene responsible for causing red blood cells to sickle back to the normal sequence. Cells capable of making red blood cells would be isolated from an individual with sickle cell disease and the DNA would be edited so that the mutation would be reversed. The edited cells would then be injected back into the patient’s blood and allowed to produce red blood cells. Because the edited cells would now have the normal beta-globin gene sequence, these individuals could produce normal hemoglobin and therefore would have normal, healthy red blood cells [7].
The first attempt to use this approach to edit human cells took place in 2017. Some of the cells that make red blood cells (CD34+ hematopoietic pluripotent stem and progenitor cells) were isolated from the bone marrow of patients with sickle cell disease and grown in a laboratory. The mutated HbS gene that encodes hemoglobin S was edited with CRISPR-Cas9 in an attempt to correct the mutation. Examination of the CD34+ cells after editing revealed an increase in normal hemoglobin A production [2].
The fact that the cells could be edited in a laboratory to successfully produce increased levels of normal hemoglobin A raised the possibility that CRISPR-Cas9 could be used clinically to treat sickle cell disease. Even so, there were still concerns about possible side effects using this approach. One major problem was the risk of gene editing to off-target sequences: that is, the risk of Cas9 cutting the beta-globin gene at a different location than the targeted mutation, which could cause any number of unpredictable changes. These changes could be potentially harmful or even fatal. However, by 2017, scientists were confident that with greater understanding of CRISPR-Cas9’s mechanisms and the rapidly advancing technology produced, this problem could be alleviated.
Since the study using CD34+ cells was successful, and because understanding of the CRISPR-Cas9 gene editing technology was continually advancing, by 2018 scientists were ready to apply the technology in clinical trials. Since that time, several trials using CRISPR-Cas9 with the goal of curing sickle cell disease have been launched. It has been reported that a clinical trial by Graphite Bio using CRIPSR technology is planned to start in 2021. The trial will attempt to directly edit the mutation in the beta-globin gene that causes sickle cell disease [15]. Other clinical trials however have taken different approaches to treating the disease using CRISPR technology.
Some trials have attempted to treat sickle cell disease by targeting specific genes other than the mutated beta-globin gene directly. One such clinical trial, by Vertex Pharmaceuticals Incorporated in collaboration with CRISPR Therapeutics, was launched in 2018. Like the study of cultured cells described above, this trial used CRISPR-Cas9 to edit CD34+ hematopoietic stem and progenitor cells. However, rather than attempting to fix the mutated beta-globin gene, this approach attempted to produce a different, nonmutated hemoglobin gene that is present in the genome but not expressed in adults [18]. This trial also collected CD34+ hematopoietic stem and progenitor cells from the patients’ peripheral blood, rather than from their bone marrow, which is less painful and invasive to the patient.
The beta-globin gene targeted in this study is called the BCL11A gene. This gene is turned on prior to birth and produces proteins that stop fetal hemoglobin production and initiate adult hemoglobin production. Fetal hemoglobin, also known as HbF, is similar to the normal adult hemoglobin (HbA), except that it has two gamma chains instead of two beta chains. Because the primary function of HbF is to supply oxygen to a developing fetus, its structure allows it to bind oxygen more strongly than HbA so that it can more easily retrieve oxygen from the mother’s bloodstream and deliver it to the fetus through the placenta. Around the time an individual is born, HbF production is stopped, and HbA begins to be produced. The BCL11A gene plays a significant role in this process by producing a protein that works with other proteins to turn off (silence) the gamma-globin genes, thus preventing gamma-globin, and ultimately fetal hemoglobin, from being produced. It also interacts directly with the beta-globin gene to produce beta-globin, a component of adult hemoglobin [9].
It has previously been shown that red blood cells produce more fetal hemoglobin when portions of the BCL11A gene are deleted, thus blocking production of the protein it encodes. Because the gamma chains in this type of hemoglobin more effectively carry oxygen through the bloodstream, the cells are less likely to be sickled than those containing the adult hemoglobin with beta chains produced by the mutated beta-globin gene when the BCL11A gene is regularly functioning [15]. Scientists reasoned that if the BCL11A gene were silenced, more fetal hemoglobin than adult hemoglobin would be produced in red blood cells, and fewer cells would be sickled.
In the trial launched by Vertex Pharmaceuticals Incorporated and CRISPR Therapeutics in 2018, cells treated with CRISPR-Cas9 at the BCL11A genes called CTX001 were transplanted into individuals with sickle cell disease [16]. The individuals experienced an initial increase in HbF levels in over 99% of their red blood cells. Over a year later, the individuals reported back with no vaso-occlusive crises or need for blood transfusions since the CTX001 treatment. While there were side effects reported, including abdominal pain and a reduced amount of white blood cells, the overall preliminary results of this trial supported further testing with CRISPR-Cas9 technology in treating the disease [17]. The estimated completion date of the trial is in May of 2022. The participants will again be observed to determine how safe and effective the trial was overall. A second measure of HbF levels will be recorded, as well as the total number of vaso-occlusive crises participants experienced and the change in PRO throughout the duration of the trial [18]. PRO is a personal assessment of one’s health conditions without the involvement of a clinician [19].
One of the first patients to be treated with CTX001 for sickle cell disease is a woman by the name of Victoria Gray. Stem cells were taken from her blood, CRISPR was used to edit her BCL11A gene so that more fetal hemoglobin would be produced using the approach described above, and the edited cells were infused back into her blood. A year after treatment, in June 2020, her hemoglobin level was 11 g/dl. This is much healthier than 7 g/dl, her hemoglobin level before treatment, and closer to the normal, healthy range of hemoglobin levels in women, 12 to 15.5 g/dl [20]. Prior to receiving the treatment, Gray averaged at seven hospitalizations a year for her sickle cell disease. In contrast, during the first year after receiving treatment, she was never hospitalized. Since these results were so encouraging, scientists are planning to expand into later phases of clinical trials (phases II or III) that will involve far more participants with this treatment [1].
All of these gene editing discoveries are new and encouraging for those in the sickle cell disease community. Some trials have proved very hopeful for the future of treating the disease, but the technology is not yet completely mastered, and the risks of serious side effects such as off-target gene editing have not yet been determined. Because these new technological advances are being designed for the benefit of those involved in the sickle cell disease community, their input is vital, and studies are being done to better understand their perspectives. One example of this is an initiative launched by the National Human Genome Research Institute in January 2020 which evaluates the perspectives of sickle cell disease patients, their family members, and their healthcare providers on incorporating CRISPR technology into sickle cell disease treatment [21]. The general consensus of those affected by sickle cell disease is optimism due to this new favorable possibility, but caution because of the potential risks and side effects [22].
The early studies described here strongly suggest that CRISPR-Cas9 could represent a major step forward in treating sickle cell disease. The current hesitation among members of the sickle cell disease community is likely to diminish as scientists gain further understanding of the technology and the genetic factors that come into play with the disease. Research in this field is progressing daily, and current clinical trials are very promising, and represent a major advance over the treatments for sickle cell disease that are presently available. It is likely that significant development in this technology will occur over the next few years, bringing hope to the millions who are struggling with the disease. It seems likely that, in addition to sickle cell disease, this approach may be used to treat many other genetic diseases as well.
Contacts: karis.weisgerber@howardcc.edu, kjones@howardcc.edu
References
[1] R. Plater. “First Person Treated for Sickle Cell Disease with CRISPR Is Doing Well.” Healthline.com. https://www.healthline.com/health-news/first-person-treated-for-sickle-cell-disease-with-crispr-is-doing-well#The-next-steps. (accessed Nov. 2020).
[2] W. Jianguo, T. Wenjing, H. Suyang, and Youli, Z, “Cellular function reinstitution of offspring red blood cells cloned from the sickle cell disease patient blood post CRISPR genome editing,” J Hematol Oncol. vol. 10, no. 1, p. 119, June 2017, doi: 10.1186/s13045-017-0489-9
[3] A. Mangla, M. Ehsan, and S. Maruvada, “Sickle Cell Anemia,” in StatPearls [Internet], Treasure Islant (FL): StatPearls Publishing, 2021.
[4] “Red blood cells – multiple sickle cells.” MedlinePlus. https://medlineplus.gov/ency/imagepages/1223.htm. (accessed Nov. 2020).
[5] A. Hamosh, “OMIM Entry – # 603903 – SICKLE CELL ANEMIA,” Online Mendelian Inheritance in Man, vol. 1, no. 603903, Sept. 2019.
[6] J. E. Maakaron. “Sickle Cell Anemia Clinical Presentation: History, Physical Examination.” Medscape.Com. https://emedicine.medscape.com/article/205926-clinical. (accessed Oct. 2020).
[7] S. Demerci, N. Uchida, and J. F. Tisdale, “Gene Therapy for Sickle Cell Disease: An Update,” Cytotherapy, vol. 20, no. 7, pp. 899-910, Jul. 2019, doi: 10.1016/j.jcyt.2018.04.003
[8] E. Smith. “Gene Therapy, From Idea to Reality.” https://lakartidningen.se/klinik-och-vetenskap-1/artiklar-1/klinisk-oversikt/2017/12/genterapi-fran-ide-till-verklighet/. (accessed Dec. 2020).
[9] V. G. Sankaran and S. H. Orkin, “The Switch from Fetal to Adult Hemoglobin,” Cold Spring Harb Perspect Med., vol. 3, no. 1, p. a011643, Jan. 2013, doi: 10.1101/cshperspect.a011643
[10] F. Uddin, C. M. Rudin, and T. Sen, “CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future,” Front Oncol., vol. 10, p. 1387, Aug. 2020, doi: 10.3389/fonc.2020.01387
[11] A. Loureiro, and G. Jorge da Silva, “CRISPR-Cas: Converting A Bacterial Defense Mechanism into A State-of-the-Art Genetic Manipulation Tool,” Antibiotics (Basel)., vol. 8, no. 1, p. 18, Feb. 2019, doi: 10.3390/antiobiotics8010018
[12] L. Banyuls et al., “In Vitro Genome Editing Using CRISPR/Cas9 to Edit SERPINA1 PiZ Mutation,” European Respiratory Journal, vol. 54, p. 5411, Sept. 2019, doi: 10.1183/13993003.congress-2019.PA5411
[13] F. A. Khan et al., “CRISPR/Cas9 therapeutics: a cure for cancer and other genetic diseases,” Oncotarget., vol. 7, no. 32, pp 52541-52552, Aug. 2016, doi: 10.18632/oncotarget.9646
[14] “Henceforth CRISPR,” Nat Biomed Eng., vol. 4, no. 11, p. 1118, Oct. 2020, doi: 10.1038/s41551-020-00646-0
[15] R. Cross. “CRISPR start-up Graphite Bio launches with $45 million for targeted DNA integration.” https://cen.acs.org/pharmaceuticals/gene-therapy/CRISPR-startGraphite-Bio-launches-45/98/web/2020/09. c&en.acs.org. (accessed Dec. 2020).
[16] “CRISPR Therapeutics and Vertex Announce Progress in Clinical Development Programs for the Investigational CRISPR/Cas9 Gene-Editing Therapy CTX001.” Sec.Gov. https://www.sec.gov/Archives/edgar/data/1674416/000119312519049058/d712065dex991.htm. (accessed Nov. 2020).
[17] H. Frangoul, et al., “CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia,” New England Journal of Medicine, vol. 384, pp. 252–260, Jan. 2021, doi: 10.1056/NEJMoa2031054
[18] “A Safety and Efficacy Study Evaluating CTX001 in Subjects with Severe Sickle Cell.” ClinicalTrials.Gov. https://clinicaltrials.gov/ct2/show/NCT03745287?term=CTX001&draw=2&rank=2. (accessed Nov. 2020).
[19] A. T. Farrell et al., “End points for sickle cell disease clinical trials: patient-reported outcomes, pain, and the brain,” Blood Adv, vol. 3, no. 23, pp. 3982-4001, Dec. 2019, doi: 10.1182/bloodadvances.2019000882
[20] “Hemoglobin test.” Mayoclinic.org. https://www.mayoclinic.org/tests-procedures/hemoglobin-test/about/pac-20385075 (accessed Feb. 2021).
[21] “Examining the Knowledge, Attitudes, and Beliefs of Sickle Cell Disease Patients, Parents of Patients with Sickle Cell Disease, and Providers Towards the Integration of CRISPR in Clinical Care.” ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/record/NCT03167450?term=crispr&cond=Sickle+Cell+Disease&draw=2&rank=. (accessed Nov. 2020).
[22]A. Persaud, S. Desine, K. Blizinsky, and V. L. Bonham, “A CRISPR focus on attitudes and beliefs toward somatic genome editing from stakeholders within the sickle cell disease community,” Genetics in Medicine, vol. 21, no. 8, pp. 1726-1734, Dec. 2018, doi: 10.1038/s41436-01801409-6
